Jama Software is always on the lookout for news on our customers that would benefit and inform our industry partners. As such, we’ve curated a series of customer spotlight articles that we found insightful. In this blog post, we share content, sourced from Mass Device, about one of our customers, Surgalign titled “FDA Clears Surgalign’s Cortera Spinal Fixation System” – which was originally published on August 24, 2022, by Sean Whooley.
FDA Clears Surgalign’s Cortera Spinal Fixation System
Surgalign (Nasdaq:SRGA) has announced that it received FDA 510(k) clearance for its Cortera spinal fixation system.
Deerfield, Illinois-based Surgalign said in a news release that the new flagship Cortera product represents a key product portfolio piece. Surgalign officials see Cotera driving the company’s future growth. It could ensure market gains in the posterior fixation market.
“The Cortera system is a testament to the spine engineering talent and expertise we’ve assembled in very short order, as we moved from zero engineers in the United States following the RTI divestiture two years ago, to approximately 30 today,” said Terry Rich, president and CEO of Surgalign. “Thanks to our team and incredible surgeon partners, we progressed from company inception to FDA 510(k) clearance with a very polished system in approximately 16 months. We are excited with the prospects the Cortera system brings to Surgalign, and those around the world who rely on our technology to drive better patient outcomes.”
Cortera, a 5.5/6mm rod pedicle screw system, offers both open and minimally invasive surgery (MIS) modules, plus a feature-rich screw design with a comparatively low profile and newly designed locking mechanism.
Surgalign designed Cortera to maximize adoption in the spine market, both today and in the future with evolving techniques and technologies. The company added that Cortera demonstrates the ways in which spinal implants will be deployed with technologies like its own HOLO Portal surgical guidance platform.
The company plans to integrate Cortera with HOLO Portal to create what it labeled “an unrivaled user experience for pedicle screw navigation.” Surgalign also has plans for additional implants and instruments to add to the system over the next few years to expand applications into a majority of posterior fixation spinal procedures.
Surgalign will offer Cortera in a limited market release, which it expects to positively contribute to its 2022 fourth-quarter results and in the coming years.
“The system is hands down the most precise, elegant and comprehensive screw that currently exists in my opinion,” said Dr. Jeremy Smith, chief of spine, Hoag Orthopedic Institute. “I find the system has an evolved sophistication that provides a high-quality user experience and enhanced clinical performance in challenging pathologies.”
Are you a Jama Software customer looking to fill open positions at your organization with prospects who have Jama Connect experience? We’d love to help! Tag us on LinkedIn (@jamasoftware) with your job posting and we’ll share it!
Incorporating Risk Traceability into Manufacturing Production Software and Preparing for the Transition from CSV to CSA
Intro
Over the years, the burden of Computer Systems Validation (CSV) has resulted in medical device manufacturers avoiding implementation of automated manufacturing production systems or upgrading long-outdated versions of software. As part of the FDA’s ‘Case for Quality’ initiative in 2011 to study quality best practice in medical device manufacturing, the FDA found that the burden of compliance, such what is expected for Computer Systems Validation (CSV), deterred technology investments and as a result, inhibited quality best-practice.
As an outcome, the FDA is expected to release a draft guidance in 2022 that outlines their new approach of Computer Software Assurance (CSA) for Production and Quality System Software. The goal is to improve software quality by focusing on the software’s impact to patient safety, impact to product quality, and impact to quality system integrity. Using a risk-based approach, manufacturers can spend more time testing to ensure software meets its intended use, instead of ensuring test protocols and reports are fully scripted are error free.
As the key to right-sizing CSA activities is based on risk, managing risk, and risk traceability and incorporating is a key activity. And there’s no need to wait for the publication of the FDA CSA draft guidance, medical device manufacturers can incorporate these risk management and traceability practices now in their CSV activities.
Keep reading below for best practices on how to extend risk management and risk traceability from the medical device development to your manufacturing production software used to manufacture your devices.
Main Points for How
Consider the product risks and hazards
Even though we are discussing manufacturing production software and the software used on manufacturing equipment, the first step is to consider the product risks and hazards. Start with your master list of hazards, hazardous situations, and associated harms developed during medical device development and determine where and how failures in the manufacturing production software and production equipment can result in those risks and hazards. These are the areas to consider as the highest risks to control, mitigate, and validate.
Next, perform additional risk analysis from the lens of the environment and manufacturing personnel that will operate said equipment and software. It is important to also protect the environment and prevent injury and harm to those interacting with the equipment. You don’t want the unintended release of hazardous chemical reagents or equipment fires that could have been prevented with built-in high temperature shut-down feature.
Create manufacturing production software intended uses and requirements
Just as one determines the intended use and design input requirements of a medical device, the same is to be done for the manufacturing production software and equipment. Incorporate the necessary features as manufacturing requirements (analogous to the design inputs of your medical device) that address the risks identified in the previous step.
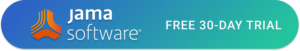
One example is based on risk of the product and demonstrates how risk traces from the development of the medical device. Consider the design of a semi-automated assembly machine for a drug-delivery device. The machine aligns and press-fits together two plastic injection-molded halves of the delivery sub-assembly. One of the main product associated risks is ensuring proper flow efficiency to ensure the proper amount of drug is delivered correctly and to the proper anatomical location.
In this case the risk traceability and translation to the assembly machine design inputs is:
Perform software testing and equipment validation based on risk
Once ready to test the production equipment software and perform equipment validation, incorporate, you guessed it, risk. All manufacturing requirements then need to be verified, however, scale the amount and type of testing for each manufacturing requirement based on the risk. Aspects of the software and equipment that are of high risk, such as preventing severe harm to operators, or the end users and patients of the medical device will have more testing and documentation than those of lower risk.
Document risk traceability of testing and equipment validation
Just as documenting risk traceability for medical device is important, it’s also important for manufacturing production software and equipment. Using a tool like Jama Connect® makes it easy to link your master list of hazards, hazardous situations, and harms to your manufacturing requirements for manufacturing production software and equipment. This increases consistency and efficiency in the risk analysis and prioritizing efforts on where it matters for patient and operator safety. The interface of Jama Connect® also makes the traceability and documentation of the associated testing and validation easy to visualize, alerting teams to gaps and incomplete testing.
Closing
Extending risk traceability from your medical device development to your manufacturing production software and equipment brings focus to what can impact patient safety and scales the testing and validation work appropriately. Incorporate risk and you’ll also be well on your way for the upcoming FDA transition from CSV to CSA.
https://www-dev.jamasoftware.com/media/2022/09/2022-09-27-preparing-for-csv-to-csa-1.jpg5121024Michelle Wu/media/jama-logo-primary.svgMichelle Wu2022-09-27 03:00:062023-01-12 16:46:20Incorporating Risk Traceability into Manufacturing Production Software and Preparing for the Transition from CSV to CSA
Jama Software is always on the lookout for news and content to benefit and inform our industry partners. As such, we’ve curated a series of articles that we found insightful. In this blog post, we share content sourced from Lifecycle Insights – Jama Connect®: Accelerating Systems Development with Requirements Management and Live Traceability™ – which was originally published on August 17, 2022, by John McMillan.
Jama Connect®: Accelerating Systems Development with Requirements Management and Live Traceability™
How does Jama Software®’s approach to managing the vast array of complex engineering requirements provide organizations a competitive advantage? Their approach is a software solution developed specifically to provide unified requirements management and traceability across organization development processes whether that be the traditional V-model, Waterfall, Agile, or otherwise. Jama Connect requirements management software with Live Traceability was designed to help development and engineering organizations improve quality, reduce rework, ensure compliance, and get high-quality complex products, systems, and software to market faster and on budget.
Historically in product development, each engineering discipline utilizes tools that are specifically suited to maximize their ability to be innovative and productive in their own design space. Communication between other disciplines and the broader organization however has historically been siloed or fragmented and often managed through “throw-over-the-wall” manual approaches. Those approaches are error-prone and often result in discovering late-stage issues downstream that result in design rework, delay product delivery, and create cost overruns that impact the organizations’ bottom line.
The Cost of Discovering Late-Stage Issues
When late-stage issues are discovered during integration testing, systems testing, and during final acceptance testing they are expensive to fix. Jama Connect platform was developed to enable organizations to detect and correct requirement and testing issues and solve disjointed discipline problems. This is provided through a requirements management platform designed specifically to help engineering organizations align people, processes, and tools in one concise application early on and throughout product development- when the cost of change is lowest.
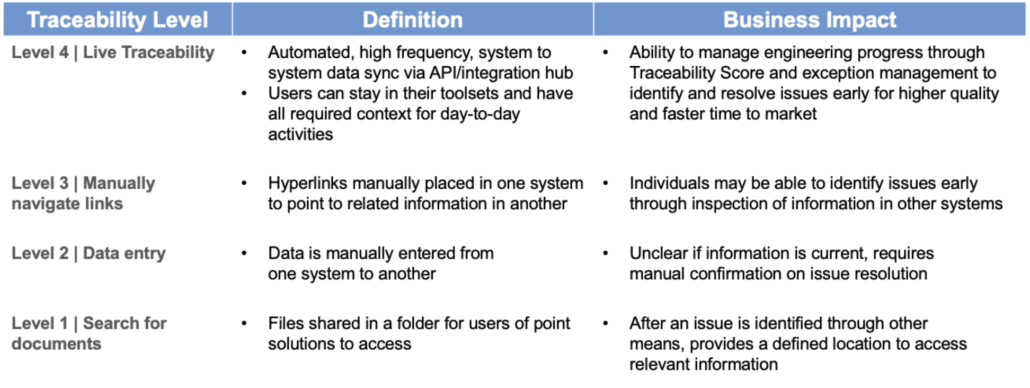
Jama Connect was designed to provide unique real-time visibility and actionable insights for the end-to-end product, systems, and software development processes. Within the application, users develop relationship models between each discipline’s tools by way of data elements. These data elements are connected with direct tool integrations with Live Traceability. Once the flow of each element’s connections is defined and reflected throughout the system – should a data element be modified, the connected element stakeholders are alerted, and each discipline can review and address any changes accordingly. The application’s unique open architecture allows integrations with a range of premium solutions across the full ALM-PLM tool ecosystem.
Jama Connects list of supported tools and plug-in integrations for Live Traceability is already quite extensive and is growing.
For Design and Simulation model-based requirements management Connect seamlessly integrates with MBSE and SysML tools including Ansys, MathWorks, Enterprise Architecture, and Catia’s No Magic.
For Task Management, Live Traceability is directly supported for Jira, Bugzilla, Azure DevOps, and TFS without any changes required by software developers’ preferred tools, methodology, or field values.
For PLM and PLE link requirements to hardware specifications and product line engineering for traceability and impact analysis are seamlessly supported for Teamcenter, Windchill, Aras, Pure-Systems, and BigLever.
For Test Automation, live trace requirements and test cases to automated testing results are supported from tools including Tricentis, Ansys, LDRA, TestRail, ZEPHYR, Vector, Jenkins, Bugzilla, and Parasoft.
For Risk Management, traceability is supported from Ansys FMEA/DFMEA calculations as well as Microsoft Excel including functions and spreadsheets, is also supported without any changes required to Risk team’s tooling or approaches.
For DevOps, Live Traceability is extended down to source code with applications including GitLab, GitHub, and Azure DevOps with no changes required to software developers’ tools or methodologies.
Though Jama Connect provides users with the ability to develop custom model system frameworks, it also includes the frameworks for plans, templates, and checklists that are specifically aligned to industry standards for medical devices, automotive, semiconductor, aerospace, defense/government, software development, and industrial manufacturing. In addition, an extensive list of industry standards and regulations are supported including ISO, IEC, FDA, EU, SEBoK, ARP, DO, and more. These industry-specific standards help organizations ensure end-to-end compliance, mitigate risk, and overall process improvement guidance.
Addressing risk management with system analysis that is tailored to each product’s industry standards and regulations, “left-shifts” risk management throughout the product’s development flow and in turn serves as an integral part of the product lifecycle process. Organizations can standardize and integrate their own risk analysis, evaluation, and risk management processes within Jama Connect’s platform to create a single source of truth for everything risk related.
In addition to risk management, Jama Connect provides critical verification and validation requirements for complex systems via test management. The tool supports customized reporting for proof of regulatory compliance and performs manual user-acceptance testing to ensure products are designed with end-users in mind. The tool generates links to disparate processes, sources, and people that increase visibility and simplifies the user’s path to compliance with traceability of tests back to its requirement. It also traces failed tests to new and existing defects for quick resolution, enabling users to reuse validated requirements saving time when testing consistent features across products.
MBSE (Model Based System Engineering) is another key area that Jama Connect provides a streamlined and collaborative data-driven approach to in the product development cycle. Jama Connect’s Companion MBSE platform combines requirements, architectures, behaviors, verification, and validation into a single model of the system by applying structure and rules for data and a consistent interface language between the parts of the system. The MBSE platform is designed to help organizations formalize the development, integration, and use of models to inform enterprise and program decision-making. It also allows non-technical stakeholders to visualize a model of the system of interest and interact with its data in familiar views like documents and spreadsheets.
A leading problem that product engineering organizations face is complying with traceability spanning siloed teams and tools (e.g., design, hardware, software, test, risk, quality) creating an increased risk of negative outcomes such as extensive rework, delays, & cost overruns. Requirements traceability across the entire systems development lifecycle is a core tenant of the systems engineering discipline and underpins industry standards to ensure higher quality, faster cycle times, and less costly rework.
RELATED
https://www-dev.jamasoftware.com/media/2022/09/2022-09-20-jama-connect-accelerating-systems-development-1.jpg5121024Decoteau Wilkerson/media/jama-logo-primary.svgDecoteau Wilkerson2022-09-20 03:00:382023-01-12 16:46:20Jama Connect®: Accelerating Systems Development with Requirements Management and Live Traceability™
This is part 2 of a two-part blog series covering our whitepaper, “The New EU In Vitro Diagnostic Regulation: What’s Changing and What You Need to Know” written by Vincent Balgos, Medial Solution Manager at Jama Software. In this paper, Vincent discusses the In Vitro Diagnostic Regulation (IVDR), developed by the EU Commission (CE), which was created to replace the previous In Vitro Diagnostic Directive (IVDD).
Part 1 of this blog series is available HERE. To read the whitepaper in its entirety, download it HERE.
The New EU In Vitro Diagnostic Regulation (IVDR): What’s Changing and What You Need to Know
The IVDR Overview
With more than 150 pages of regulations, there were many changes to strengthen and grow the path to IVDs marketed and distributed in the EU. The IVDR provides a more comprehensive approach to regulating devices as it encompasses the entire product lifecycle: from initial concept to design and manufacturing, to continual on-market support along maintaining good documentation practices. For the purpose of this paper,
only a few selected topics will be covered in detail with some additional insights from an industry perspective.
Medical companies that develop IVD’s marketed to the European Union, and its population. This includes non-EU-based companies that have products in the EU region.
What is impacted?
In-Vitro Diagnostics (IVDs) products and accessories that are used to perform tests on various sample types to help diagnose a condition, detect infections, or monitor drug responses.
When is it happening?
Date of Application: May 26, 2022, where only IVDR applications will be accepted by NB. A two-year window period afterwards to allow companies to transition their IVDD to IVDR certification. By May 2025, all IVDD certificates will be voided, with IVDR covering all placed IVDs in market.
What countries are impacted?
European Union and the United Kingdom specifically, but companies from around the world who have products in the UK or EU are also required to conform.
Why are things changing?
To improve the safety and quality of IVDs in the EU market.
Discuss on Key Topics
Changes to Classifications
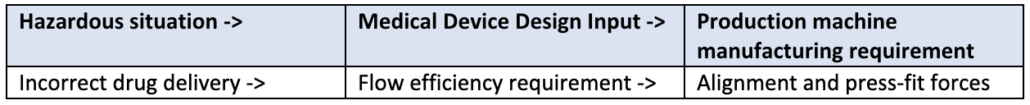
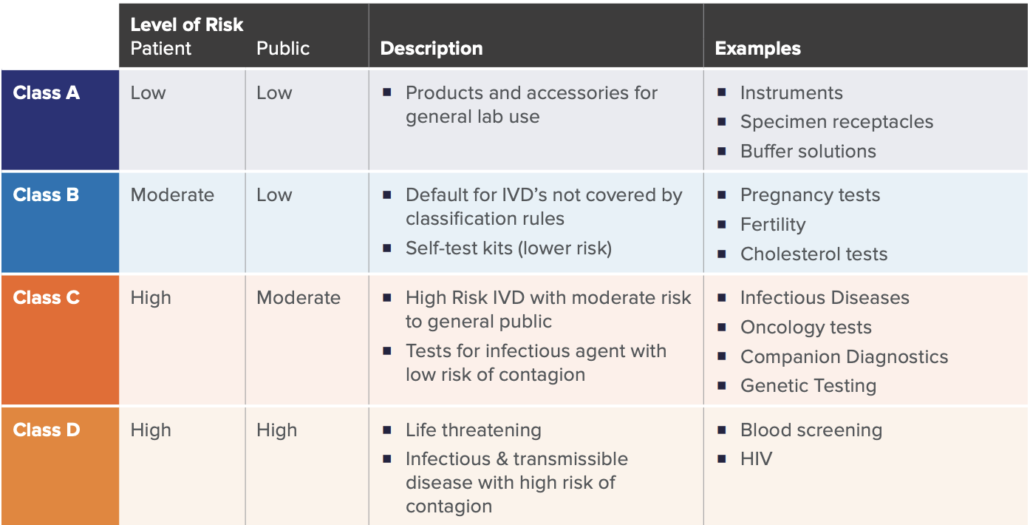
A key significant change of the new regulation is how IVD’s are classified. Similar to the previous directive, a risk-based approach (with respects to public and patient) is used to classify IVDs under the new IVDR framework. There are four main classes as listed in the table below, established by seven classifications rules defined in Annex VIII of IVDR.
While there may be nuances to the rules set, these four categories broadly cover the majority of the IVD spectrum. This new classification schema only allows Class A devices to be self-certified by manufacturers, whereas Class B, C, and D require more assessment and certification by notified bodies.
As in similar regulatory pathways, device classification is significant in determining the overall requirements, as the higher the IVD risks, the more onerous the regulatory requirements, and the higher the involvement with an external Notified Body. For example, a new HIV Test would be categorized as Class D, which would require the highest number of internal activities during design, development, on-market support, and associated documentation. This would also require the highest amount of interaction, assessment, and certification from the Notified Body. Furthermore, specific regulations such Post-Market Surveillance, Quality Management System elements, and annual updates to reports are required for higher risk class (C and D) devices.
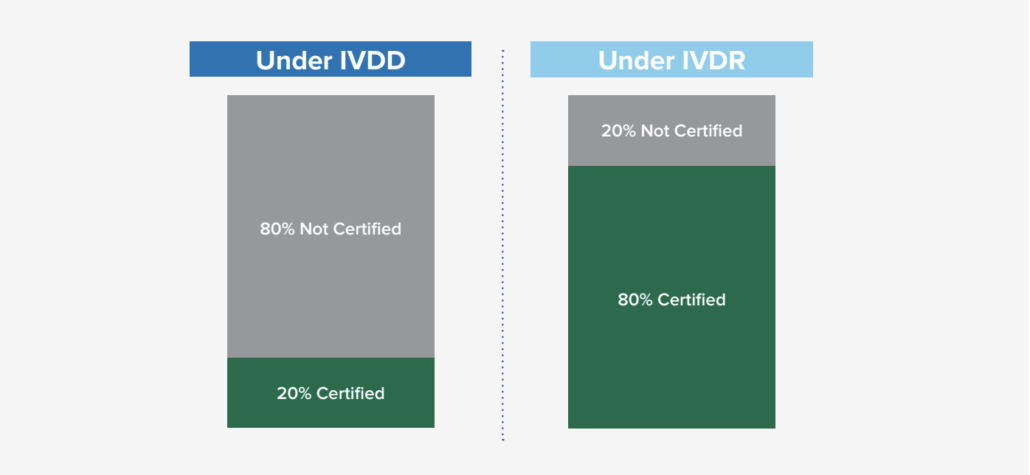
Based on general research from industry subject matter experts, it was estimated that only 20% required Notified Body certification under the previous IVDD, while 80% did not require certification. With the new classification schema and requirements under IVDR, that ratio has flipped where it is expected that the majority of IVD’s (80%) will now need some Notified Body involvement. This new shift (in engaging Notified Bodies and the new requirement) is significant in many ways as it not only impacts the manufacturers, but also the Notified Bodies as demand for their engagement has risen exponentially. There are some concerns about the current Notified Body capacity, so it is encouraged to start engaging with a Notified Body proactively, as the backlog to engage could be longer than anticipated.
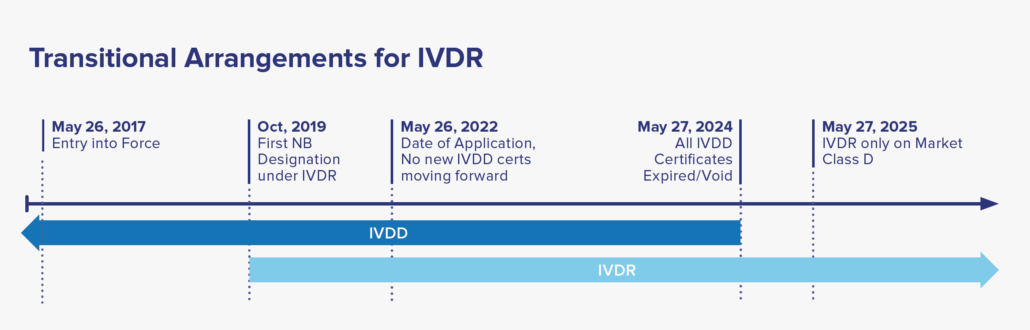
For certified IVD’s that are currently on-market classified under the IVDD guidance, reclassification to the IVDR categories and recertification to meet the IVDR is required for continual sale and distribution to EU market. Under the IVDR, there are no grandfathering clauses to allow the IVDD devices to remain on market after May 2025. Considering there are many IVD’s in use, the EU established a five-year timeline to allow manufacturers to transition to IVDR. See the timeline below for more details.
Since IVDR’s announcement in 2017, many companies and SME’s (including this author) started to update
their internal procedures, adjust development and documentation activities, and hire additional resources
in response to the impending changes. In addition, remediations to current devices’ Design History Files
(DHFs) to align with regulations were also underway. These include adding additional testing for new
requirements such as performance studies, clinical evaluations, etc. These activities may be significant,
and a major resource pull from other ongoing projects. Therefore, it’s critical to acknowledge that the new
IVDR regulations impact not only future but current IVDs on the market as well.
Under Article 15, a new IVDR requirement is that manufacturers are required to have a regulatory compliance expert in their organization to be responsible for the compliance of the in-vitro diagnostics regulations. This person must be a qualified regulatory expert with previous demonstrated qualification such as 1) formal certification from approved regulatory body and/or 2) minimum of four years of industry experience as a regulatory affairs professional in the IVD field. This role (new for some organizations) provides general regulatory affairs guidance, interpretation of regulations to internal teams, and helps facilitate discussions with Notified Bodies, regulatory agencies, and EU Competent Authorities.
Establishing Risk Management
While not a new requirement to IVD practices, Annex I Chapter I of the IVDR has multiple languages referring to and establishing risk management practices. This further substantiates the EU focus on a riskbased approach when developing devices and encourages many best practices that Jama Software® has seen many of our IVD customers follow.
This new language includes the following requirements:
To establish, implement, document, and maintain a risk management system.
To enforce continuous and iterative risk management process with regular updates to the risk files throughout the device lifecycle, especially after the product has been launched to market.
To reduce risks as far as possible without adversely affecting the benefit-risk ratio and inclusion of this analysis in technical files submission. This includes risks related to use errors of the device.
To consider design accommodations to assure that characteristics of safety and performance are maintained during the transport and storage of the product, and for the expected lifetime of the product.
To minimize all known and foreseeable risks and be acceptable when weighed against the potential benefits.
This updated language continues the industry practice of risk management that is further established in ISO 14971 “Medical Devices – Application of Risk management to Medical Devices” and TR 24971 “Medical Devices – Guidance on the application of ISO 14971.” Based on the reasons why the IVDR came into fruition (PIP accidents), it can be surmised that an organization’s risk management process will be under significant scrutiny by the Notified Body. Therefore, Risk Management Procedures have been a focal point of update for organizations to strengthen risk practices and ensure compliance. Remediation of risk files may also be warranted for devices currently on the market, or soon to be on the market in the EU.
Based on this author’s experience, this risk activity alone requires significant time and resources to accomplish. Considering some risk files could have significate number of documents (plans, evaluations, reports) with details that require comprehensive review from many stakeholders, this is an effort that needs formal organization support to successfully comply with the IVDR and its compliance timeline. Therefore, it is recommended to prioritize
appropriately and revisit the Risk Management section, and other impacted areas of the IVDR as soon as possible.
General IVDR Guidance for Medical Device Companies
Based on discussions with various IVD customers, general research, and internal experience, we recommend the following guidance:
Determine the new IVDR classification for each of your devices on market, or plan to be launched in the EU, and their associated requirements. Consult with Regulatory affairs to proactively affirm
classification with a notified body.
Review and remediate procedures and documents to include new IVDR regulation languages and requirements. Based on your organization’s level of compliance, this could be a significant activity so
may need management support.
Identify the accredited regulatory affairs expert in your organization that will be responsible to drive the activities to comply with the IVDR regulations. This may include updating general regulatory
procedures, product development processes, and for existing technical documentation.
Review and update risk management procedures to include new requirements such as regular updates of the risk files, incorporate use-risk scenarios, and ensuring the benefit-risk comply with
new language.
As with many types of changes in regulations, these have substantial impact on how organizations and their teams operate in the design, development, and manufacturing documentation of IVDs. It is encouraged to proactively review these new regulations as it may require significant time and resource to adapt to continue developing IVD’s for the European market.
DISCLAIMER
Jama Software is not an accredited regulatory subject matter expert, so these are general guidance and insights from working with many IVD customers, general research, and some internal experience. It is suggested to work with a certified Regulatory Affairs consultant for formal recommendations for your organization.
Accelerate Innovation in Medical Device Development While Adhering to Industry Regulations
With the new IVDR, it is expected that manufacturers will need to shift to a more regimented process of developing, manufacturing, and managing IVD’s. Similar to other regulatory pathways, good requirements management is the best practice in ensuring compliance with regulations, reducing risk, and launching safe and effective products.
Jama Connect® for Medical Device Development helps medical device teams reduce the effort required to achieve regulatory compliance throughout the development process. With this solution, medical device teams can manage design controls for device requirements and related risks, simplifying regulatory submissions and audit preparations while accelerating time to market. Jama Connect creates a digital thread for systems engineering and
ensures Live Traceability™ and alignment across the product development lifecycle to seamlessly connect development solutions and facilitate product success.
ABOUT THE AUTHOR, VINCENT BALGOS
Vincent Balgos currently leads the Medical Solution at Jama Software. Prior to joining Jama Software, he worked in the medical device / IVD industry for over 17 years with roles in systems engineering, product development and project management. Vincent has successful history in launching new products to the global regulated market, and is experienced in product development, risk management, quality systems, and medical device regulations.
Part 1 of this blog series is available HERE. To read the whitepaper in its entirety, download it HERE.
https://www-dev.jamasoftware.com/media/2022/09/2022-09-15-ivdr-part2_1024x512-1.jpg5121024Vincent Balgos/media/jama-logo-primary.svgVincent Balgos2022-09-15 03:00:412023-01-12 16:46:21The New EU In Vitro Diagnostic Regulation: What’s Changing and What You Need to Know: Part 2
What Is OSLC?
OSLC (Open Services for Lifecycle Collaboration) is an “open” standard designed to facilitate communication between tooling primarily used in engineering disciplines. The initial work was done by IBM in 2009 and in 2013 governance moved to OASIS. The idea behind OSLC is to provide a common layer for tool vendors so that connection between tools can be created without having to write and maintain individual connectors between each set of tools. In theory, this reduces the burden on tool vendors and gives users confidence that tools that support OSLC will interoperate without issue.
What tools support OSLC?
Many tool vendors do not directly support OSLC. Jama Software® integrates with a wide variety of best-of-breed tools and most of them do not come with native OSLC connectors. Of the 25 most prevalent tools that we see in the market, 20% have some OSLC capability, and only 16% have a company- supported connector. Contrast that with the fact that 100% of these tools have a company-supported REST API and you can clearly see the direction of the market.
OSLC services a relatively small niche that often requires consulting and some technical assistance to setup. Given that many of the tools that utilize OSLC are desktop based, the IT environment becomes a challenge for integration. This has led many to rely on third-party vendors to broker the OSLC integration layer and provide the support necessary. At Jama Software we have partnered with MiD and their product Smartfacts to enable this connection. Jama Connect® has a robust REST API and through our partnership with MiD we can support those customers who need an OSLC integration, while extending our integration capabilities to the much wider systems engineering ecosystem.
Using the integration hierarchy to achieve Live Traceability™
Most companies who are considering an OSLC integration are doing so to improve requirements traceability across their product development process. Requirements traceability approaches range from rudimentary, manual approaches, to automated synchronization across tools. At Jama Software we’re focused on helping our customers achieve Live Traceability™ via automated synchronizations of best-of-breed tools to a common traceability model. For those desktop tools that do not fully support automated synchronization, a lower level of traceability is the best that can be achieved.
Organizations that are heavily invested in a specific toolchain suite can leverage OSLC to reduce the interop challenge inherent in suites that have been created through acquisition. Embedding user interfaces into other applications and maintaining links allows a user to navigate through the “suite” without having to open each application. When dealing with desktop applications, this can reduce the need to load up each tool individually. It is certainly better than trying to sync information through manual efforts.
In desktop tool scenarios and with tools that do not have a web API, OSLC might be as far as you can go. In which case, Jama Software has you covered. For those who want to move beyond the single user context and achieve Live Traceability, we recommend Level 4 integrations. Utilizing easy-to-implement, universal web standards we’ve helped hundreds of organizations achieve the user-level benefits of contextual data while also elevating the systems engineer’s visibility to the process and organization level.
Contact Jama Software today to find out how we can help you with OSLC and further you on your journey towards Live Traceability.
https://www-dev.jamasoftware.com/media/2022/09/2022-09-13-oslc-what-is-it-1.jpg5121024Josh Turpen/media/jama-logo-primary.svgJosh Turpen2022-09-13 03:00:032023-01-12 16:46:21OSLC – What Is It and What Are Its Challenges?
Jama Software is always on the lookout for news and content to benefit and inform our industry partners. As such, we’ve curated a series of articles that we found insightful. In this blog post, we share content sourced from Medtech Insight – European Regulatory Roundup, July 2022: Threat Of Ongoing Hurdles Masks Progress – which was originally published on August 1, 2022, by Amanda Maxwell.
European Regulatory Roundup, July 2022: Threat Of Ongoing Hurdles Masks Progress
Executive Summary
The first seven months of 2022 have seen significant progress on documents, tools and new structures needed for the MDR and IVDR implementation. But with major hurdles ahead, in July it was still the problems and not the successes taking center stage.
A significant amount of progress has been made in finalizing documents and processes needed for the implementation of the Medical Device Regulation (MDR) and IVD Regulation (IVDR), as the European Commission’s latest update to its rolling plan, issued in July, showed.
But despite this headway, the outcome of a recent extensive survey by MedTech Europe “clearly indicates an urgent need for immediate action” by decision-makers to help keep needed medical devices available in Europe.
In the analysis of its survey results, MedTech Europe warns that unless swift action is taken, the clinical benefits of new and improved device designs will be more likely to become available to patients in other markets ahead of the EU, requiring patients in Europe to wait until the MDR system is ready.
Indeed, MDR hurdles are deterring some 50% of companies from prioritizing the EU as their launch market, the survey found, and 33% of respondents’ medical devices are currently planned for discontinuation.
The outcomes highlight the lack of responsiveness from notified bodies, unpredictable certification time and non-harmonized interpretations of the same requirements of the MDR, not only among notified bodies as a group but also within them individually.
The urgency of addressing the EU’s regulatory problems was also emphasized by industry association, COCIR, during July. In its paper, the body proposes a series of detailed solutions that it believes would prevent anticipated bottlenecks at notified bodies. Included among its proposals are to: expand the use of remote audits to all products generally, without the need for specific justifications; and allow notified bodies more flexibility in defining the appropriate technical documentation sampling (e.g. lower the number of technical files based on risk class and not groups).
Another July reader favorite on the topic of avoiding a scenario where uncertified devices have to be unnecessarily removed from the market was a Medtech Insight interview with Bassil Akra, CEO of consultancy firm Akra, published at the end of June.
He wants regulators to acknowledge that notified bodies are receiving a huge number of applications and have not been able to accept them all because of the magnitude of work involved in dealing with them in the context of new regulations.
Moreover, he said, some manufacturers have not been able to even apply to a notified body yet because their notified body has not been designated and/or others offering services in their area have no capacity.
The Spanish Agency for Medicines and Healthcare Products’ Medical Devices Certification Division was the only new notified body designation in July. This is the first and likely only Spanish notified body under the MDR.
This brings the total number of EU MDR notified bodies to 31, while those designated in the context of the IVDR remain at seven.
Analysis was published during the month highlighting the challenges that manufacturers face in finding a notified body active in their particular product area. It emerged that only two of the 31 notified bodies designated under the MDR are authorized to carry out conformity assessments under all MDR product codes. Four out of the seven designated under the IVDR have full authorization under that regulation.
Class D is the highest risk category under the new IVDR, and legacy IVD Class D must be in full compliance with the new rules by 26 May 2025. Common specifications are like standards but more technically detailed. In this case, they focus particularly on IVD performance characteristics.
New products had to meet the requirements by 26 May this year, and these missing common specifications have been causing challenges and confusion among manufacturers and notified bodies.
Also in the areas of diagnostics, the European Medicines Agency issued in July final guidance outlining the procedure that IVDR-designated notified bodies must follow when seeking a scientific opinion on the suitability of a companion diagnostic (CDx) with the concerned medicinal product.
Eudamed
Delays and uncertainty over the launch of the updated version of the Eudamed medical device database, an MDR and IVDR cornerstone, have caused medtech stakeholders much frustration. In July, however, there was more certainty when the commission published an updated timeline.
The commission expects to declare all six modules of the updated Eudamed medical device database to be sufficiently ready to allow the launch of the full system in the second quarter of 2024.
This will then be followed by two critical deadlines:
Six months after the publication of the notice, i.e., Q4 2024, the requirements of Eudamed will become mandatory for the following modules: actor registration; vigilance; clinical investigation and performance studies; and market surveillance.
24 months after the publication of the notice, i.e., Q2 2026, Eudamed will become mandatory for the following modules: UDI/device registration; notified bodies and certificates.
Also in July, new EU guidance was published to help IVD manufacturers understand in detail what they should do pending the full launch of the database.
This is not the first text of its kind that has been made available. In May 2021, the month that the MDR fully applied, a similar document was published in the context of the MDR.
In the UK, meanwhile, there have been fears that the stormy political waters following Boris Johnson’s resignation as prime minister could impact the timing of the publication of the new, post-Brexit UK medtech regulations. But with new prime minister due to be in place by 5 September, and unlikely to deviate much from the plans of their predecessor, the hiatus is likely to be short.
In the meantime, it has emerged that device manufacturers will have a clearer picture of the post-Brexit system of medtech regulation in the UK once a number of focus groups, to be appointed by the Medicines and Healthcare products regulatory Agency (MHRA), can focus on developing key aspects of guidance that will accompany the new regulations. This step follows publication of the response to the UK consultation on the future shape on the new regulations.
https://www-dev.jamasoftware.com/media/2022/09/2022-09-08-EU-roundup-1-1.jpg5121024Jama Software/media/jama-logo-primary.svgJama Software2022-09-08 03:00:142023-01-12 16:46:22European Regulatory Roundup, July 2022: Threat Of Ongoing Hurdles Masks Progress
This is part 1 of a two-part blog series covering our whitepaper, “The New EU In Vitro Diagnostic Regulation: What’s Changing and What You Need to Know” written by Vincent Balgos, Medial Solution Manager at Jama Software. In this paper, Vincent discusses the In Vitro Diagnostic Regulation (IVDR), developed by the EU Commission (CE), which was created to replace the previous In Vitro Diagnostic Directive (IVDD).
We will share the link to part 2 when it publishes. In the meantime, you can download the eBook HERE.
The New EU In Vitro Diagnostic Regulation (IVDR): What’s Changing and What You Need to Know
Learn more about how IVDR differs from IVDD, key takeaways from the new regulation, and guidance for how to adapt.
Disclaimer: The IVDR regulation is broad and requires focused review and interpretation by each organization — so by no means is this paper intended to be all-exclusive, as it will only discuss select topics.
Jama Software® is not an accredited regulatory body, so these are general discussions and insights from our experience working with many IVD customers, general research, and some internal subject matter expertise. It is suggested to work with a certified Regulatory Affairs consultant (a new IVDR regulation) to obtain formal recommendations for your organization.
If you’re looking for guidance on who to work with when it comes to regulatory compliance, TÜV SÜD has provided certification and testing services for manufacturers and suppliers of medical devices and in vitro diagnostics for over 30 years.
They combine expert medical product testing knowledge with a global network of internationally accredited laboratories and facilities, providing you with a one-stop solution. In fact, Jama Connect® is certified through TÜV SÜD as a software tool for development of medical devices according to IEC 62304.
Introduction
In May of 2022 a paradigm shift emerged in how In-Vitro Diagnostics (IVD’s) will be developed, managed, and regulated in the European Union (EU). The EU Commission (CE) has developed new regulations named the In Vitro Diagnostic Regulation (IVDR) to replace the previous In Vitro Diagnostic Directive (IVDD). The main goal of the IVDR is to improve upon the quality, safety, and reliability of IVD’s within the European market. This will change the current status quo as IVDR has been predicted to have a significant impact in medical device organizations with IVD sale and business operations.
In this whitepaper, we will provide an overview of the new regulation, discuss some specific topics, and offer considerations for organizations as they adapt to this new paradigm.
Overview of the IVDR and The Significant Impact on EU
Figure 1. CE Mark
Prior to the IVDR, the In-Vitro Diagnostics Directive (IVDD) was the governing regulation for devices placed in Europe. Officially adopted in 1997, the IVDD established the regulatory requirements for CE Marking approval for in vitro diagnostics¹. In order to sell and market IVD’s in the European Union, manufacturers need to show compliance with the essential requirements prior to marking the product with the CE label. The CE mark allows for legal distribution
of the IVD within the European Economic Area (EEA)². The CE mark indicates conformity across many different types of products and is based on compliance with specific European regulations based on the product type. See example marking to the left.
For medical devices and IVDs, compliance to the EU Medical Device Directive (MDD) and the EU IVDD was required to obtain CE marking, respectively.
IVDD was established in 1997 by the EU for trade within the EEA with 27 EU members plus Iceland, Liechtenstein, and Norway.
IVDD applies to all Reagents, Calibrators, Kit, Instrument, Equipment, Systems used for in vitro diagnostics purposes in the EEA, regardless of origin of design and manufacturing.
Essential Requirements included requirements for design, production, labeling, and the instructions for use (IFU). Some specific requirements included the diagnostic’s analytical sensitivity and specificity, accuracy, repeatability, and reproducibility.
There are four general categories that are based on level of risk to public health and/or patient.
Annex II List A – Highest risk which require notified body review including HxV’s such as HIV, HBV, HCV
Annex II List B – Moderate Risk including IVD’s such as HLA, Glucose monitoring
Self-Test – Examples include pregnancy home tests, and cholesterol
General – No notified body required as OEM can ‘self-declare’ conformity
Key factors such as the device classification, risk level to patients/public, etc. would determine the manufacturer’s level of approach to developing, manufacturing, and documenting the IVD. A common industry practice for launching an IVD to the global market was that organizations would first launch their products in the EU, and then to broader markets. Due to less rigidity of the IVDD when compared to other countries, it was easier, faster, and more economical for companies to launch there first. The involvement of an external notified body was also less rigid, so many organizations tended to follow the least resistant pathway to market, with many following the ‘self-declaration’/ self-certification pathway. Learnings from the EU launch (e.g., clinical studies) could then be leveraged when then submitting to the more rigid regulatory pathways such as the U.S. Food and Drug Administration (FDA).
This common approach enabled organizations to get new products to the market faster through the regulatory pathway. In the author’s experience, this approach was practiced on many of the IVD’s developed throughout their career in multiple diagnostic applications. The general regulatory roadmap was to have initial launches in EU markets and then proceed with FDA pathway. This provided additional time to work on FDA submission activities since the level of rigidity and documentation was expected to be much higher. However, with the IVDR enforcement now in full effect, it is expected to have a tectonic shift in how manufacturers develop IVDs.
As seen with many types of general regulations, changes are commonly in response to mass incidents, generally with negative impact resulting in patient injury and sometimes even death. The US FDA has seen their regulations shift in reaction to mass incidents including the Therac-25 (radiation therapy) and Dalkon Shield (intrauterine device). The accidents led to significant legislative changes to prevent recurrences and improve industry practices to ensure ‘safe and effective’ products.
The emergence of IVDR follows a similar path, where there were European several high-profile events that led to the regulation update. The most notable was the Poly Implant
Prosthesis (PIP) breast implant scandal (based in France) that impacted many patients with high incidents of ruptured implants with unapproved industrial silicone filling. You can read more about the incident here, and the subsequent
clinical recommendations here.
This event led to significant updates to the medical device space with the culmination of the EU Medical Device Regulation published in 2017. Following the MDR initiative, the incumbent IVDD was also overhauled into the new IVDR paradigm which entered into force on May 26, 2017
Stay tuned for Part 2 of this blog series. To read the whitepaper in its entirety, download it HERE.
https://www-dev.jamasoftware.com/media/2022/08/2022-09-01-ivdr-part1_1024x512-1-1.jpg5121024Vincent Balgos/media/jama-logo-primary.svgVincent Balgos2022-09-01 03:00:412023-01-12 16:46:24The New EU In Vitro Diagnostic Regulation: What’s Changing and What You Need to Know: Part 1
Jama Software is always on the lookout for news on our customers that would benefit and inform our industry partners. As such, we’ve curated a series of customer spotlight articles that we found insightful. In this blog post, we share content, sourced from Times Aerospace, about one of our customers, SITA titled “SITA unveils eVISA and ETA to transform borders” – which was originally published on July 28, 2022.
SITA unveils eVISA and ETA to transform borders
SITA has launched SITA eVisa and SITA Electronic Travel Authorisation to meet the growing demand from governments for digital visa systems to stimulate national economies after COVID-19.
Governments globally are shifting to modern travel authorization solutions, like electronic visas and Electronic Travel Authorisations (ETAs). According to the World Travel & Tourism Council (WTTC), traditional visas – applications made via a consulate or embassy – decreased from 77% in 2008 to 53% in 2018. There is a growing demand for digital travel solutions.
The advantages of digital authorization solutions include improved security, reduced administrative burden, easier travel, and increased visitor flows, promoting spending that benefits local economies and creates employment. For example, one government’s introduction of an eVisa scheme covering 40 plus countries in 2014-2015 led to a 21% increase in international visitor arrivals and the creation of 800,000 jobs accounted for around 20% of the growth seen in the country’s travel and tourism over the period.
The mobile capability of SITA’s new eVisa and ETA capability allows travelers to make applications and provide their biometric information using their personal devices before they travel.
SITA’s eVisa and ETA solutions provide visas containing ICAO’s Visible Digital Seal (VDS), an encrypted bar code that enables visas and ETAs, paper or electronic, to be digitally verified for authenticity, offering enhanced security and fraud prevention.
Jeremy Springall, Head of SITA AT BORDERS, said: “Adopting eVisa and ETA supports national prosperity. We’ve productized our proven and robust travel authorization systems to benefit more nations around the world as they shift to digitalize and future-proof their borders. The solutions help countries to cope with growing passenger volumes, improve security and efficiency, and deliver a more seamless travel experience that travelers demand, removing the complexities of applying for traditional visas”.
Springall added: “The adaptability of these two solutions means that they are fully interoperable with existing border control and airline systems. And, they comply with international standards and best practices.”
RELATED
https://www-dev.jamasoftware.com/media/2022/08/New-SITA.png5121024Decoteau Wilkerson/media/jama-logo-primary.svgDecoteau Wilkerson2022-08-31 03:00:432023-10-25 12:32:49SITA Unveils eVISA and ETA to Transform Borders
Systems Engineering for Product Development with Live Traceability
Complexity of Product Development is Exploding
In today’s world, technological innovations across embedded systems, the Internet of Things (IoT), and artificial intelligence (AI) are aggressively pushing the envelope on product complexity and accelerating digital transformation. The next generation of autonomous cars, electric vehicles, robots, and space shuttles are all complex systems that need to seamlessly interact with multiple layers of software, hardware, and humans across different environments.
Because complex technology products require intricate interfaces with various systems or sub systems, there is an inherent challenge of coordinating the multiple development teams, who are often geographically spread across the world. Furthermore, large teams typically work on different stages of product development within their own silos. The different development tools, architecture, and IT environments used across various teams and organizations can significantly add to the challenges of developing new technologies.
Manufacturing sectors will reap the AI technology benefits of enhanced monitoring and supply chain optimization. In financial institutions, AI techniques can be used to identify which transactions are likely to be fraudulent. Through AI, the industry could also adopt fast and accurate credit scoring while automating manually intense data management tasks. Disruptions in the transport and logistics realm could include autonomous trucking and delivery on the roads and automated picking in warehouses.
Most complex system development starts with defining user needs and requirements, which involves design, software, and hardware teams developing deliverables that ultimately get tested and integrated for delivering the product. Given the disparate nature of these teams, there is an inherent need to optimize and continuously keep track of systems information from concept to development and delivery. However, there is often a gap in terms of how information is consumed across engineering departments, as well as its availability and traceability. The digital thread in engineering today is still a collection of different tools, which means traceability is usually accomplished in a cumbersome and manual fashion via time-consuming meetings and disconnected documents.
Challenges in Traceability
Traceability is a key both to product development and software engineering for tracking the systems development lifecycle. Helpful in achieving regulatory compliance, traceability can also be used within software engineering for running test cases and gaining valuable insights.
While some organizations make it a point to map product development stages and improve traceability to gain these benefits, traceability is often considered after-the-fact by others. This means that once an issue is identified, there is effort required to trace back where the error originally occurred to prevent that same failure from occurring in the future. This approach can cause multiple cost overruns, product delays, and inefficient management of change impacts. Think of addressing a car recall or fixing a complex medical device or industrial equipment— the costs can be enormous. By emphasizing the importance of traceability from the start, organizations can avoid the extra effort and cost associated with implementing it after an error has been made.
The solution lies in bringing a practical approach of Live Traceability™ to the systems engineering world. Optimizing and measuring the systems development process with Live Traceability is a crucial competitive advantage that companies can leverage. For any complex product, systems, or software development, the requirements and user needs form the first level of abstraction. Live Traceability can be measured and monitored by using systems requirements as an anchor to manage information used in different stages of the systems development lifecycle.
At Jama Software, we define live requirements traceability as the ability for any engineer at any time to see the most up-to-date and complete upstream and downstream information for any requirement— no matter the stage of systems development or how many siloed tools and teams it spans. Live Traceability of system requirements is required by industry standards to ensure product safety and forms the foundation for digital engineering and model-based systems engineering. This enables the engineering process to be managed through data and its performance improved in real time.
Through better requirements management and a focus on traceability, companies can improve their systems development performance with higher quality products and improved cycle times. The industry is now embracing the idea that to improve engineering efficiency within the systems development process, we need to continuously monitor and measure traceability.
https://www-dev.jamasoftware.com/media/2022/08/2022-08-02-IEEE-v2-managing-complexity-systems-engineering-1.jpg5121024Nikhil Rai/media/jama-logo-primary.svgNikhil Rai2022-08-17 03:00:412023-01-12 16:46:26Managing Complexity in Systems Engineering for Product Development with Live Traceability
Launching spacecraft and satellites into orbit are highly complicated endeavors and like many large organizations, this rocket and spacecraft manufacturer was struggling to provide visibility, manage, changes to requirements, and meet aggressive schedules.
In 2010, this industry-leading space exploration technology company selected Jama Connect to help its team develop new ways of working to simplify requirements management, improve visibility, and customer communication, and create an efficient, paperless product delivery process.
They saw significant improvements to its requirements management process with Jama Connect, including:
Reducing meeting times, simplifying cumbersome specification docs, and improving overall communication to ensure all requirements were accounted for accurately
Better traceability from requirements to derived requirements to verification events
Improved customer relations with shared project visibility and enhanced communications
Jama Connect® helps an innovative aerospace company keep track of thousands of requirements by providing a platform for greater visibility and collaboration with its team and customers
This innovative aerospace launch company designs, manufactures, and launches the world’s most advanced rockets and spacecraft. In just ten years, the company has emerged as one of the fastest growing aerospace companies in the world working toward their vision to revolutionize space travel.
CHALLENGES
• Providing visibility and managing customer expectations
• Managing inevitable change to requirements and other important details
• Eliminating rework in order to meet aggressive schedules
EVALUATION
• Utilize Jama Connect’s Review Center for open customer and internal communication
• Track requirements for faster change management
• Maintain common requirements in one place to prevent repeats
RESULTS
• Helped reduce meeting times, simplify cumbersome specification docs, and improve overall communication to ensure all requirements were accounted for accurately
• Provided a method for traceability from requirements to derived requirements to verification events
• Improved customer relations with shared project visibility and enhanced communications
• Accounted for contract milestones and ensured timely payments
Launching spacecraft and satellites into space is a highly complicated endeavor. Like many other large organizations tackling complex projects, the company initially faced a few critical business challenges that Jama Connect could help them address:
• Providing visibility and managing customer expectations
• Managing inevitable change to requirements and other important details
• Eliminating rework in order to meet aggressive schedules
As a customer, NASA needed visibility into the company’s progress to ensure key requirements were being met. Prior to using Jama Connect, the company did not have an easy way to share quick status reports on requirements verification. As a result, many hours were spent compiling status and change-control reports.
This process, although effective, took too much time. Hours were spent in meetings reviewing large cumbersome spreadsheets – going line by line to review thousands of details. The company had to implement complex macros to pull information from Microsoft Excel into Word documents to create specific reports. This was time-consuming for everyone involved. The company wanted to share real-time information with NASA for their review and approval, but they did not have an easy way to accomplish this level of collaboration prior to using Jama Connect.
Solution: One Small Step for the Company, One Giant Leap for Efficiency
The company implemented Jama Connect in 2010, wanting to develop new ways of working to simplify requirements management, improve visibility and customer communication, and create an efficient, paperless product delivery process.
https://www-dev.jamasoftware.com/media/2022/08/2022-08-16-customer-story-innovative-aerospace-manufacturer-chooses-jama-connect-1-1.jpg5121024Decoteau Wilkerson/media/jama-logo-primary.svgDecoteau Wilkerson2022-08-16 03:00:182023-01-12 16:46:27Innovative Aerospace Manufacturer Chooses Jama Connect® to Help Revolutionize Space Transportation




 Since IVDR’s announcement in 2017, many companies and SME’s (including this author) started to update
Since IVDR’s announcement in 2017, many companies and SME’s (including this author) started to update